海綿状脳症(プリオン病)は、病的形態のプリオンタンパク質が発達に関与している疾患です。私たちはプリオン病についてますます知っていますが、重要な側面は不明のままです-現在、医学はこれらの疾患から患者を治療する手段を持っていません。
海綿状脳症、またはプリオン病は、一生の間に発症する可能性がありますが、他の人は出生から存在する遺伝性の遺伝子変異から発生します。このグループにはいくつかの人間の実体があり、クロイツフェルトヤコブ病や致命的な家族性不眠症などがあります。
プリオン病は長い間非常に神秘的です。細菌、ウイルス、真菌などの他の病原体とは異なり、それらは核酸を含んでいません-プリオンはタンパク質のみで作られています。プリオン病の理論はS.プルシナーによって発見され、この発見は科学界で高く評価されました-1997年に研究者はノーベル医学賞を受賞しました。プリオンの概念が生まれてからかなり長い年月が経過しましたが、一部の科学者はまだそれが不完全であると信じており、これらの状態の性質をさらに調査しています-しかし、海綿状脳症の原因となるいくつかの要因はすでに確認されています。
プリオン病:原因
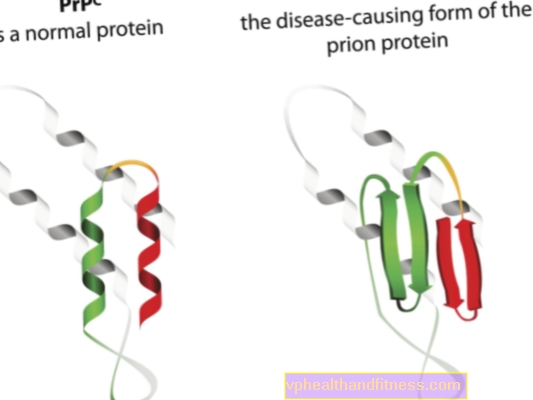
プリオン病の病因は、正常なプリオンタンパク質の病原性、病原性形態への変換に関連しています。プリオンは、すべての人間の体に見られるタンパク質分子です。それらの機能はまだ完全には解明されていませんが、通常の条件下では、プリオンタンパク質は体に害を与えないことが知られています。ただし、プリオンの構造が変化して病原性粒子になると、いくつかの海綿状脳症の1つが発症します。体内で自然に発生するプリオンはPRPCと呼ばれ、異常な形態はPRPSCと呼ばれます。後者は、神経組織に沈着物の形で蓄積して損傷を与える可能性があるだけでなく、正常なプリオンを奇形の形に変換する能力があるため、深刻な問題です(簡単に言えば、PRPSC病原性の可能性がある正常なタンパク質に「感染」することができます)。
また読む:ハンチントン病(ハンチントン舞踏病):原因、症状、治療筋震え-原因。筋肉振戦とはどういう意味ですか?最も速く死ぬ病気:ショック、エボラ、ダム、攻撃、緊急[GALE ...基本的に、海綿状脳症には3つの原因があります。
- 散発性(病原性変異は体細胞で発生し、患者の人生中に発生する)、
- 家族(両親から受け継がれた突然変異の負担から生じる)、
- 通路(これらの粒子で汚染された成長ホルモン製剤や、海綿状脳症を患っている人からの角膜移植などによる、人体への病原性プリオンの導入に関連する)。
海綿状脳症:クロイツフェルト・ヤコブ病
クロイツフェルトヤコブ病(CJD)は、1920年代初頭に初めて説明されました。病気には4つのタイプがあります:
- 散発的なCJD(すべてのCJDケースの最大9/10を占める最も一般的なもの)
- CJDの故郷
- ベルト付きCJD
- CJDのバリアント
クロイツフェルト・ヤコブ病のさまざまな変種の経過における臨床像は、変動する可能性があります。この海綿状脳症のグループの過程で最も一般的な病気は次のとおりです。
- 認知症障害(記憶、注意および集中力の進行性の悪化を含む)
- ミオクローヌス(突然の筋肉のジャークのような不随意運動)
- 小脳機能障害(例えば、平衡障害により顕在化する)
- ぼやけた視界
- 錐体および錐体外路症状
CJDバリアントの過程で、精神障害(不安、抑うつ気分など)、痛み、および上記以外の不随意運動も現れることがあります。
クロイツフェルトヤコブ病の予後はよくありません。たとえば、散発的なCJDの患者では、疾患の症状が発現してから死亡するまでに平均4〜5か月かかります。
海綿状脳症:Gerstmann-Straussler-Scheinker症候群
Gerstmann-Straussler-Scheinker症候群(GSS)は、通常、家族で発症し、PRNP遺伝子の遺伝性変異によって引き起こされます。これは、最も進行が遅い海綿状脳症であると考えられています。 GSSチームには次のものが含まれます。
- 脊髄小脳失調症
- 構音障害
- 認知症障害
- 嚥下障害
- 眼振
- 筋肉の緊張の増加
GSSと診断された患者の時間は変動し、一部の患者では発症から10年以上経過して死亡します。
海綿状脳症:致命的な家族性不眠症
致命的な家族性不眠症は、PRNP遺伝子の変異によって引き起こされるプリオン病です。この病気は非常にまれで、これまでに世界中の28家族で診断されています。致命的な家族性不眠症の過程で、最初の症状は睡眠不能です。この問題は不安障害と幻覚を経験している患者をもたらします。夜間休息がない状態が続くことの影響は、自律神経系の機能不全(心臓機能の変化、発汗、消化器系疾患を含む)であり、体重の漸進的な減少もあります。致命的な家族性不眠症のより進行した段階では、ホルモン障害が現れ、認知症の症状が病気の過程で発生します。
他の海綿状脳症と同様に、致命的な家族性不眠症の予後は不良です。通常、患者は発症から3年以内に死亡します。
海綿状脳症:プロテアーゼに対する感受性が変動するプリオノパシー
説明されている海綿状脳症の発生は、主にPRNP遺伝子の変異に関連しています。ただし、これらの変異はこの遺伝子の異なるコドンに関係するため、いくつかの異なるプリオン病が区別されます。比較的最近(2008年に)記述されたユニットは、プロテアーゼに対する感受性が変動するプリオノパシーです。この病気に苦しんでいる人々は、PRNP遺伝子の3つものコドンに変異を持っています。
可変プロテアーゼ感受性を伴うプリオノパシーでは、患者は以下を経験します:
- 認識機能障害
- 精神疾患の極度の重症度:それらは陶酔感と興奮である可能性がありますが、重大な無関心でもあります
- 構音障害
- 失語症(言語障害)
このプリオノパシーにおける疾患の平均期間は4年未満です。
海綿状脳症:クル
現在、クルはほとんど存在しない病気と見なされています。これは、人食い行動を実践したパプアニューギニアの部族の代表者から発見されました。この海綿状脳症の主な症状は、進行性小脳運動失調です。それは不随意運動(主に舞踏病、振戦およびアテトーゼの形で)、ならびに尿失禁および便失禁を伴うことがある。クルの患者はまた、気分が大きく変動し、原始的な反射(例えばしゃぶり)を発症します。このプリオン病の場合の非常に特徴的な問題は、泣いたり笑ったりすることを強いられます-後者の現象のため、クルは「笑う死」と呼ばれることがあります。
海綿状脳症:診断
プリオン病は、患者の症状に基づいて疑われる可能性があります。しかし、それらはプリオンに関連しない他の多くの病気の過程でも現れるかもしれないので、それらは全く非特異的です。このため、海綿状脳症の診断には以下も使用されます:
- 画像検査(例:プリオンタンパク質による脳の変性に関連する変化を検出できる磁気共鳴画像)、
- 実験室試験(例えば、MAP-タウ、S-100または14-3-3タンパク質などの脳脊髄液中のタンパク質濃度の評価)、
- 遺伝子検査(患者の突然変異の存在を検出するため)、
- 免疫組織化学的検査(プリオンタンパク質に対する抗体を使用)。
診断は脳の剖検によっても確認でき、海綿状脳症に特徴的な変化を見つけることができます。これらは、さまざまな分布の異なる構造の海綿状病変(特定の疾患の実体に応じて)、アミロイドプラーク、およびニューロンの欠陥である可能性があります。
海綿状脳症:治療
プリオン病は現在不治である-長年にわたって行われている多くの研究にもかかわらず、医学には、その進行を遅くしたり、完全に阻害する可能性のある薬物がまだありません。対症療法は、海綿状脳症の患者に使用されます。その目的は、症状の強度を緩和し、生活の質を可能な限り改善することです。
しかしながら、海綿状脳症の治療に関する研究はまだ進行中です。科学者たちはさまざまな方法を使おうとしています-最初の例は遺伝子治療です。それらは核酸とそれらの構造に存在する突然変異に影響を及ぼします-遺伝子治療を適用する目的は、遺伝暗号の誤りを無力化することです。別のアプローチが免疫療法の基礎です-病原性プリオンを排除する役割を持つ抗体を作成する作業が進行中です。海綿状脳症と戦う可能性があると考えられる別の方法は、一旦患者の体内に導入されると、病理学的タンパク質を中和する合成タンパク質分子の使用による治療である。
おすすめ記事:
脳症-原因、種類、症状