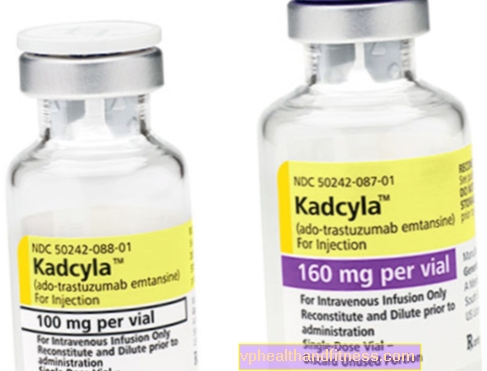
1つのバイアルには、再構成用の100 mg(160 mg)の粉末が含まれています。再構成後の注入用に、20 mg / mlの濃度のトラスツズマブエンタンシン濃縮物5 ml(8 ml)が含まれています。
| 名前 | パッケージの内容 | 活性物質 | 価格100% | 最終更新日 |
| カドシラ | 1バイアル、調製用粉末最終的解決infに。 | トラスツズマブエンタンシン | 2019-04-05 |
アクション
抗がん剤。トラスツズマブエンタンシンは、安定したMCCチオエーテルリンク(4- シクロヘキサン-1-カルボキシレート)を介してDM1(メイタンシン誘導体、微小管阻害剤)に共有結合したヒト化抗HER2 IgG1モノクローナル抗体であるトラスツズマブを含む抗体薬物複合体です。 。 EmtansineはMCC-DM1複合体です。 DM1とトラスツズマブのカップリングは、HER2を過剰発現している腫瘍細胞に対する細胞毒性薬の選択的作用を引き起こし、腫瘍細胞におけるDM1の細胞内濃度を直接増加させます。 HER2に結合すると、トラスツズマブエンタンシンは受容体を介して内在化され、続いてリソソーム分解が起こり、DM1を含む異化産物(主にリジン-MCC-DM1)が放出されます。トラスツズマブエンタンシンの作用機序は、トラスツズマブとDM1の活性によって媒介されます。トラスツズマブエンタンシンは、トラスツズマブと同様に、受容体の細胞外ドメイン(ECD)のドメインIV、Fcγ受容体および補体C1qに結合します。さらに、HER2受容体のECDドメインの活性を阻害し、ホスファチジルイノシトール3-キナーゼ(PI3-K)経路のシグナル伝達を阻害し、HER2を過剰発現するヒト乳癌細胞における抗体依存性細胞傷害(ADCC)を媒介します。 DM1はチューブリンに結合します。チューブリン重合を阻害することにより、DM1とトラスツズマブエンタンシンの両方が細胞周期のG2 / M期で細胞を停止させ、最終的にはアポトーシスによる細胞死を引き起こします。 MCCリンカーは、全身のDM1放出を減らし、標的部位でのその濃度を高めます。トラスツズマブエンタンシンは脱共役され、細胞リソソーム内でタンパク質分解により異化されます。 DM1は主にCYP3A4によって代謝され、CYP3A5によってより少ない程度に代謝されます。トラスツズマブのT0.5は約4日です。 3週間間隔で複数回の静脈内注入を行った後、トラスツズマブエンタンシンの蓄積はありませんでした。年齢はトラスツズマブエンタンシンの薬物動態に影響を与えませんでした。
投与量
静脈内。準備は医師によって処方され、癌患者の治療に経験のある医療専門家の監督の下で行われるべきです。トラスツズマブエンタンシンを投与されている患者は、HER2陽性の癌-免疫組織化学(IHC)スコア3+またはin situハイブリダイゼーション(ISH)で比率≥2を持つ必要があります。テストは、CEマーキングの付いた体外診断(IVD)テストを使用して実行する必要があります。 CE IVDテストが利用できない場合、テストは別の検証済みテストで実行する必要があります。医療ミスを避けるために、バイアルのラベルをチェックして、準備および投与されている薬がカセプラ(トラスツズマブエンタンシン)であり、ハーセプチン(トラスツズマブ)ではないことを確認することが重要です。推奨用量は3.6 mg / kg体重です。 3週間ごとに点滴静注(21日周期)。患者は腫瘍の進行または許容できない毒性が達成されるまで治療されるべきである。開始用量は、90分の静脈内注入として投与する必要があります。注射部位は、投与中の薬物の皮下浸透の可能性があるため、注意深く監視する必要があります。以前の注入が十分に許容されていた場合、その後の投与は30分の注入として与えられるかもしれません。注入中およびその後少なくとも30分間は患者を観察する必要があります。患者が輸液に関連する症状を経験した場合は、輸液率を減らすか中断する必要があります。生命にかかわる注入反応が起こった場合は、トラスツズマブエンタンシンを中止する必要があります。アレルギー/アナフィラキシー輸液反応用の薬や緊急用機器は、すぐに使用できるように用意しておく必要があります。計画された用量を逃した場合は、できるだけ早く投与する必要があります。次のサイクルまで待たないでください。投与スケジュールは、3週間の投与間隔を維持するように調整する必要があります。次の投与量は、推奨投与量に従って投与する必要があります。症候性の副作用の管理には、断続的な中止、用量の減少、または治療の中止が含まれる場合があります。減量スケジュール(開始用量3.6 mg / kg):最初の減量3 mg / kg; 2回目の減量2.4 mg / kg bw;さらに減量が必要な場合は、治療を中止する必要があります。 ASTおよびALT標高の線量変更ガイドライン:グレード2(> 2.5から≤5x ULN)-線量調整は必要ありません。グレード3(> 5から≤20x ULN)-AST / ALTがグレード≤2(> 2.5から20 x ULN)に減少した場合は準備を使用します-治療を中止します。高ビリルビン血症の用量変更規則:グレード2(> 1.5から≤3x ULN)-ビリルビンレベルがグレード≤1に低下したときに準備を使用します。 (> ULNから1.5 x ULN)、線量調整は不要。グレード3(> 3〜≤10x ULN)-ビリルビン濃度がグレード1以下(> ULNから1.5 x ULN)に減少する場合は、製剤を使用してから、用量を減らします(用量削減スケジュールを参照)。グレード4(> 10 x ULN)-末端処理。血小板減少症の用量変更規則:グレード3(血小板濃度25,000から3)-血小板濃度が1以下の場合に製剤を使用します。 (例:≥75,000/ mm3);線量調整は必要ありません。グレード4(血小板濃度3)-血小板濃度が1以下になったときに製剤を使用します。 (例:≥75,000/ mm3)その後、線量を減らします(線量削減スケジュールを参照)。心室機能不全患者における用量変更の原則:LVEF 45%-治療を継続する。 LVEF 40%から≤45%で、同時に駆出率が減少します-プレパレーションを使用しないでください。 3週間以内にLVEFを再評価します。LVEFがベースラインからまだ10パーセントポイント以上の場合は、治療を中止します。症候性CHF-治療を中止する。小児および青年:播種性乳がん(MBC)はこの集団では知られていないため、18歳未満の子供および青年における安全性と有効性は確立されていません。患者の特別なグループ。グレード3または4の末梢神経障害を発症した患者では、グレード≤2に達するまで治療を一時的に中断する必要があります。治療を再開するときは、減量スケジュールで概説されているように、減量を検討することができます。 65歳以上の患者では、用量調整は必要ありません。 75歳以上の患者の治療の安全性と有効性を判断するには、データが不十分です。軽度または中等度の腎障害のある患者では、最初の用量調整は必要ありません。重度の腎不全患者における用量調整の潜在的な必要性は決定できないため、これらの患者は注意深く監視する必要があります。軽度または中等度の肝障害のある患者では、最初の用量調整は必要ありません。トラスツズマブエンタンシンは、重度の肝障害のある患者では研究されていません。治療中に観察された既知の肝毒性のために肝障害のある患者を治療するときは注意が必要です。与える方法。製剤は、医療関係者によって溶解および希釈され、静脈内注入として投与される必要があります。ボーラスまたは急速注射として投与してはなりません。
適応症
トラスツズマブとタキサンを併用または個別に以前に治療された、HER2陽性で手術不能な局所進行または転移性乳がんの成人患者の単独療法。以前に局所進行または全身性疾患の治療を受けた患者、またはアジュバント療法の完了中または完了後6か月以内に再発した患者。
禁忌
活性物質または製剤の他の成分に対する過敏症。
予防
肺炎を含む間質性肺疾患(ILD)の症例は、トラスツズマブエンタンシンの臨床試験で報告されています。一部は急性呼吸不全に関連しているか、致命的でした。 ILDまたは肺炎と診断された患者には、製剤による治療の中止が推奨されます。進行したがんおよび関連疾患の合併症に関連する安静時呼吸困難の患者は、肺合併症を発症するリスクが高い可能性があります。製剤による治療中に、肝毒性、肝臓の結節性再生肥大(NRH)や薬物誘発性肝障害による死亡など、肝臓と胆道の重篤な障害が観察されました。肝毒性の可能性があることが知られている併存症および/または併用薬も影響を受けた可能性がある。肝機能は、治療開始前とその後の投与前に監視する必要があります。ベースラインのALT上昇(例:肝転移に関連)のある患者は、肝不全を発症する素因があり、グレード3〜5の肝毒性または肝検査室レベルの上昇のリスクが高くなります。 NRHの症例は、肝生検を行うことによって診断されました。 NRHの発生は、門脈圧亢進症の臨床兆候および/または肝CTで見られる肝硬変のような画像を持っているが、血清トランスアミナーゼ値は正常で、他の肝硬変の証拠がないすべての患者で考慮する必要があります。 NRHと診断された場合は、製剤による治療を中止する必要があります。治療開始前の血清トランスアミナーゼが2.5 x ULN(正常値の上限)を超える患者、または総ビリルビンが1.5 x ULNを超える患者では、研究されていません。血清トランスアミナーゼ活性> 3 x ULNで、同時に総ビリルビン濃度> 2 x ULNの場合、治療を終了する必要があります。肝障害のある患者を治療するときは注意が必要です。治療中に左心室機能不全のリスクが高まります。トラスツズマブエンタンシンで治療された患者では、左心室駆出率(LVEF)の低下が50年で観察され、初期のLVEF値は低かった(25 kg / m2)。標準的な心機能検査(心エコー図またはマルチゲートラジオアイソトープ血管造影)は、治療の開始前に、定期的に(たとえば3か月ごとに)実行する必要があります。ベースライン時の患者のLVEFは、ほとんどの臨床試験で50%以上でした。うっ血性心不全、治療を必要とする重度の不整脈、心筋梗塞の既往歴、またはランダム化前の6か月以内の不安定狭心症、または進行がんによる安静時呼吸困難のある患者は、研究から除外されました。左心室障害の場合は、次の投与を延期するか、治療を中止する必要があります。輸液反応によりトラスツズマブ治療を中止した患者におけるトラスツズマブエンタンシン療法の効果は研究されていません。この患者グループでは、この薬による治療は推奨されません。症状が解消するまで、輸液関連の重篤な反応を示す患者では、治療を中断する必要があります。反応の重症度の臨床的判断に基づいて、治療の再開を検討する必要があります。生命にかかわる注入反応が発生した場合は、治療を中止する必要があります。過敏症のためにトラスツズマブを中止した患者におけるトラスツズマブエンタンシン治療の効果は研究されていません。これらの患者におけるトラスツズマブエンタンシンの使用は推奨されません。患者は、注入関連の反応と同じ症状である可能性のある過敏症/アレルギー反応について監視されるべきです。重度のアナフィラキシー反応が観察されています。過敏症(その後の注入中に反応が増加する)が発生した場合は、トラスツズマブエンタンシン治療を中止する必要があります。出血事象(CNS、呼吸器、胃腸出血を含む)のリスクがあるため、抗凝固剤または抗血小板薬と併用する場合は注意が必要であり、追加のモニタリングを検討する必要があります。トラスツズマブエンタンシンの各用量を投与する前に、血小板数を監視することをお勧めします。血小板減少症(≤100,000/ mm3)の患者と抗凝固療法を受けている患者(たとえば、ワルファリン、ヘパリン、低分子量ヘパリン)は、トラスツズマブエンタンシンによる治療中は注意深く監視する必要があります。トラスツズマブエンタンシンは、治療開始前の血小板数が100,000 / mm3以下の患者では研究されていません。血小板減少症がグレード3以上(3)に悪化した場合は、毒性がグレード1(75,000 / mm3以上)になるまで使用しないでください。トラスツズマブエンタンシンを用いた臨床試験では、グレード1の末梢神経障害、主に感覚障害が発生しています。 Grade 3以上の末梢神経障害の患者は、臨床試験への参加から除外されました。グレード3または4の末梢神経障害の患者の治療は、合併症がグレード≤2に減少するまで定期的に中断する必要があります。患者は神経毒性の兆候がないか定期的に監視されるべきです。生物学的製剤のトレーサビリティを向上させるために、投与された薬物の商品名を患者ファイルに明確に記載(または記載)する必要があります。 18歳未満の子供および青年における薬物の安全性と有効性は、この集団では播種性乳がんが見つからないため確立されていません。ボーラスまたは急速注射として投与してはなりません。
望ましくない活動
非常に一般的:尿路感染症、血小板減少症、貧血、低カリウム血症、不眠症、末梢神経障害、頭痛、出血、鼻出血、咳、呼吸困難、口内炎、下痢、嘔吐、吐き気、便秘、口渇、腹痛、発疹、筋骨格痛、関節痛、筋肉痛、疲労、発熱、無力症、悪寒、トランスアミナーゼの増加。共通:好中球減少症、白血球減少症、過敏症、めまい、味覚異常、記憶障害、ドライアイ症候群、結膜炎、視覚障害、流涙の増加、左心室機能不全、高血圧、消化不良、歯肉出血、皮膚掻痒症、脱毛症、爪の病気、手足足底紅斑感覚異常、じんま疹、末梢性浮腫、アルカリ性ホスファターゼの増加、注入に関連する反応(皮膚の発赤、悪寒、発熱、呼吸困難、低血圧、喘鳴、気管支痙攣、頻脈)。珍しい:肺炎(ILD)、肝毒性、肝不全、結節性再生性過形成、門脈圧亢進症、注射部位の血管外漏出(紅斑、圧痛、皮膚の刺激、痛み、腫れ)。さらに、高ビリルビン血症が観察されています。臨床試験では、血清トランスアミナーゼ(グレード1〜4)の上昇は、トラスツズマブエンタンシンによる治療中に発生し、一般に一過性でした。トランスアミナーゼの上昇はほとんどの場合一時的であり、投与後8日でピークに達しました。その後、毒性はグレード1に減少するか、次のサイクルの前に解消しました。累積的な影響もありました(グレード1-2のALT / AST上昇の患者の割合は、その後のサイクルで増加しました)。トランスアミナーゼ値が上昇した患者では、患者の大多数が、トラスツズマブエンタンシンの最終投与から30日以内にグレード1の毒性の低下または回復を経験しました。左心室機能障害は、臨床試験に参加している患者の2.2%で見つかりました。ほとんどの場合、グレード1または2のLVEFの低下は無症候性であり、グレード3または4の毒性は、通常、治療の最初のサイクル中に患者の0.4%で観察されました(1–2)。出血性事象の重篤なエピソード(グレード≥3)が全患者の2.2%で報告されました。血小板減少症は、臨床試験の患者の24.9%で発生し、治療の中止につながる最も一般的な副作用でした。臨床試験では、血小板減少症の発生率と重症度はアジア人の患者で高かった。これらの合併症を発症した一部の患者は、抗凝固療法も受けました。致命的な出血エピソードと重度の出血合併症があり、抗トラスツズマブエンタンシン抗体が陽性であると診断された患者の5.3%が出血しました。
妊娠と授乳
妊婦へのトラスツズマブエンタンシンの使用は推奨されません。妊娠する前に、女性は胎児に害を及ぼす可能性があることを知らされるべきです。ただし、妊娠した場合は直ちに医師の診察を受けてください。妊娠中の女性がトラスツズマブエンタンシンで治療されている場合は、集学的チームによる綿密なモニタリングが推奨されます。トラスツズマブを妊娠中の女性に投与すると、胎児への危害または死亡を引き起こす可能性があります。準備で扱われた妊婦の子供において、羊水過少症のケース、それらのいくつかは致命的な肺低形成に至ったことが報告されました。トラスツズマブエンタンシンの細胞毒性成分であるDM1は催奇形性があり、胚毒性を示す可能性があります。女性は、トラスツズマブエンタンシン療法を開始する前に母乳育児を中止する必要があります。患者は、治療を中止してから7か月後に母乳育児を開始する場合があります。妊娠の可能性がある女性は、薬物による治療中、およびトラスツズマブエンタンシンの最終投与後7か月間、効果的な避妊を使用する必要があります。避妊の効果的な方法は、男性または女性のパートナーも使用する必要があります。
コメント
輸液関連の反応を経験している患者は、これらの症状が解決するまで、機械を運転したり使用したりしないでください。に基づいて作成された情報のSPC 2017年7月13日現在のSmPCはwww.roche.plで入手できます。
相互作用
ヒト肝ミクロソームでの代謝研究のin vitro結果は、DM1が主に酵素CYP3A4によって代謝され、CYP3A5によってより少ない程度に代謝されることを示しています。 CYP3A4の強力な阻害剤(ケトコナゾール、イトラコナゾール、クラリスロマイシン、アタザナビル、インジナビル、ネファゾドン、ネルフィナビル、リトナビル、サキナビル、テリスロマイシン、ボリコナゾールなど)の併用は、DM1レベルと毒性を高める可能性があるため避けてください。 CYP3A4を阻害しないか、ごくわずかしか阻害しない代替製剤を検討する必要があります。強力なCYP3A4阻害剤の併用が避けられない場合、可能であれば、CYP3A4阻害剤が循環から排除されるまで(約3阻害剤の半減期)トラスツズマブエンタンシンの投与を遅らせることを検討してください。ただし、強力なCYP3A4阻害剤を併用し、トラスツズマブエンタンシンによる治療を遅らせることができない場合は、そのような場合の副作用について患者を注意深く監視する必要があります。
準備には物質が含まれています:トラスツズマブエンタンシン
償還された薬物:いいえ